Research
Unified Workflow for the Rapid and In-Depth Characterization of Bacterial Proteomes

Miriam Abele, Doctoral candidate
Bacteria are the most abundant and diverse organisms among the kingdoms of life. Due to this excessive variance, finding a unified, comprehensive, and safe workflow for quantitative bacterial proteomics is challenging. In this study, we have systematically evaluated and optimized sample preparation, mass spectrometric data acquisition, and data analysis strategies in bacterial proteomics. We investigated workflow performances on six representative species with highly different physiologic properties to mimic bacterial diversity. The best sample preparation strategy was a cell lysis protocol in 100% trifluoroacetic acid followed by an in-solution digest. Peptides were separated on a 30-min linear microflow liquid chromatography gradient and analyzed in data-independent acquisition mode. Data analysis was performed with DIA-NN using a predicted spectral library. Performance was evaluated according to the number of identified proteins, quantitative precision, throughput, costs, and biological safety. With this rapid workflow, over 40% of all encoded genes were detected per bacterial species. We demonstrated the general applicability of our workflow on a set of 23 taxonomically and physiologically diverse bacterial species. We could confidently identify over 45,000 proteins in the combined dataset, of which 30,000 have not been experimentally validated before. Our work thereby provides a valuable resource for the microbial scientific community. Finally, we grew Escherichia coli and Bacillus cereus in replicates under 12 different cultivation conditions to demonstrate the high-throughput suitability of the workflow. The proteomic workflow we present in this manuscript does not require any specialized equipment or commercial software and can be easily applied by other laboratories to support and accelerate the proteomic exploration of the bacterial kingdom.
PUBLICATIONS:
Abele, M., Doll, E., Bayer, F. P., Meng, C., Lomp, N., Neuhaus, K., Scherer, S., Kuster, B., & Ludwig, C. (2023). Unified Workflow for the Rapid and In-Depth Characterization of Bacterial Proteomes. Mol Cell Proteomics, 22(8), 100612. https://doi.org/10.1016/j.mcpro.2023.100612
Plant Peptidomics: Unveiling Bioactive Responses to Bacterial Stress
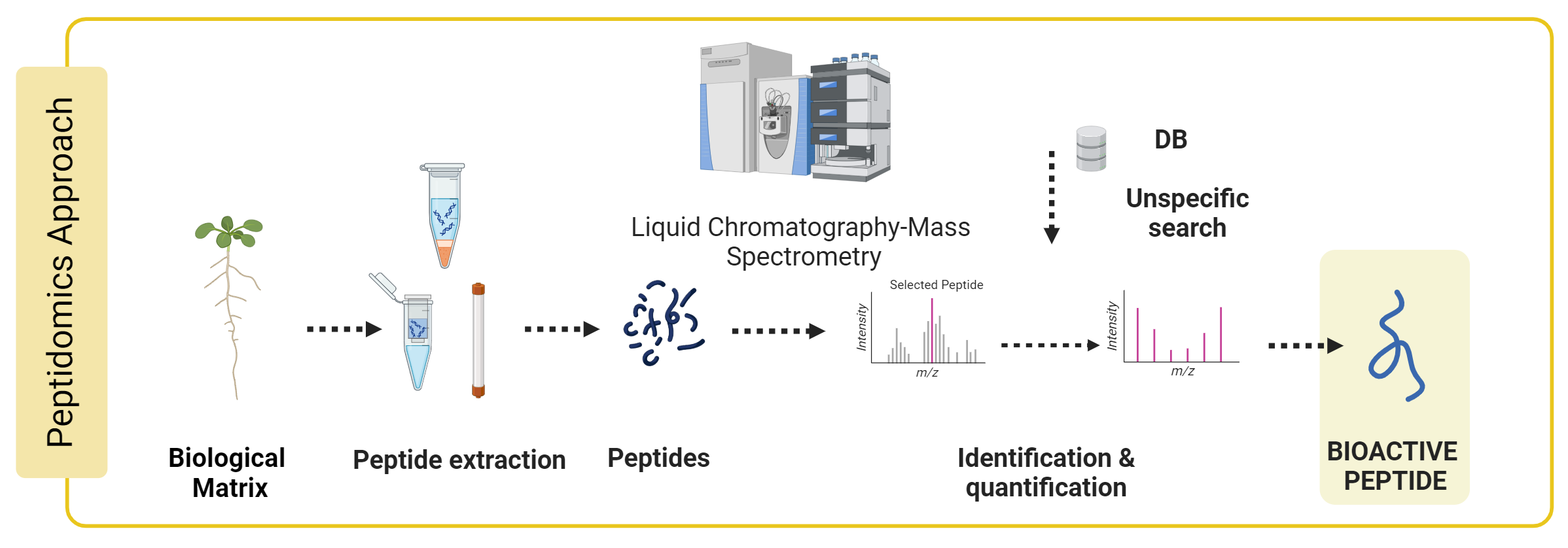
Genc Haljiti, Doctoral candidate
Scientific question
All organisms consist of bioactive peptides essential to numerous physiological functions. Bioactive peptides discovered in plants, such as phytocytokines and peptide hormones, are crucial to abiotic stress management and pathogen defense. Given that sample preparation and data evaluation present unique analytical challenges, the molecular investigation of bioactive peptides has attracted relatively little attention in the field of proteomics to date.
Approach
This work aims to establish a peptidomics workflow suited for various plant species and sample types, such as tissues and apoplastic wash fluid (AWF). To accomplish this, new mass spectrometric analysis methods and peptide extraction techniques must be established to interpret different bioactive peptides.
Expected results
The established peptidomics workflow will be applied to various biological questions, including how the plant peptidome responds to bacteria, handles abiotic stress, or during germination.